第一作者:Ming-Hua Li,Shuo Wang
通讯作者:胡劲松
通讯单位:中国科学院大学
研究亮点:
1. 开发了一种利用氢键促进的二甲基铵萃取策略。
2. 制备湿度和温度范围窗口显著扩展。
3. 无掺杂的聚(3-己基噻吩)(P3HT)基CsPbI3钙钛矿太阳能电池效率
达到20.25%,并表现出表现出卓越的湿度和操作稳定性。
一、无机钙钛矿存在的问题与挑战
尽管有机-无机混合钙钛矿太阳能电池(PSCs)取得了高达26.1%的认证效率,但有机成分在潮湿、光照和高温条件下表现不稳定。通过用无机铯离子替代有机阳离子,从根本上提高了内在稳定性,为制备稳定的PSCs提供了极大的希望。然而,常用的DMAPbI3(二甲基铵[DMA])或“HPbI3”辅助结晶法制备CsPbI3薄膜往往会导致DMAPbI3残留,从而降低光伏性能和稳定性。
二、成果简介
有鉴于此,中国科学院大学胡劲松教授团队开发了一种通用的氢键促进DMA萃取策略来制备高质量的γ-CsPbI3没有DMAPbI3的薄膜残留。这种对环境无害的结晶过程显著延长了制造湿度和温度窗口,提高了器件稳定性。不含掺杂剂的聚(3-己基噻吩)(P3HT)的PSC 可达到20.25%的高效率,并具有出色的储存和照明稳定性。
本研究EQE采用Enlitech- QE-R3011产品进行测量。
图片
三、结果与讨论
要点1:PAA作用下DMAPbI3辅助CsPbI3结晶动力学工程
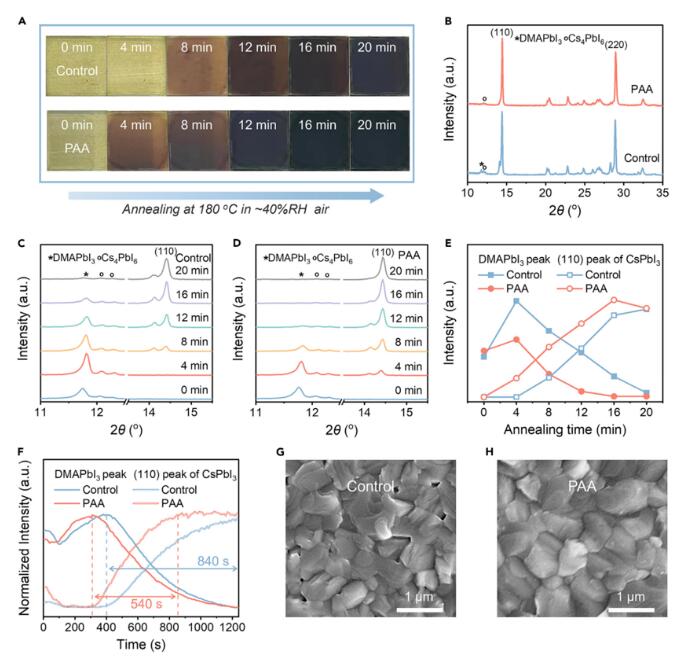
首先作者将一定量的PAA溶解在DMF溶液中,添加到CsPbI3前驱体中,用于制备经PAA处理的薄膜。纯CsPbI3和经PAA处理的CsPbI3分别被标记为对照和PAA样品。如图1A所示,与对照样品相比,PAA样品表现出从淡黄色到深黑色的颜色变化更快。使用X射线衍射(XRD)技术研究了晶体结构和相变过程所得到的钙钛矿薄膜(图1B)与标准的γ-CsPbI3相相匹配。PAA薄膜的(002)峰的消失(图S2)表明PAA处理有助于形成更倾向于(110)取向的结构。值得注意的是,对照样品中仍存在一个约11.8°的小峰,该峰对应DMAPbI3。此外,两种钙钛矿薄膜都显示出弱的Cs4PbI6衍射峰,有助于稳定晶格结构。进一步通过时间依赖的XRD研究了PAA处理对DMAPbI3辅助CsPbI3结晶过程的影响。
图1 CsPbI3的结晶动力学研究
进一步通过时间依赖的XRD研究了PAA处理对DMAPbI3辅助CsPbI3结晶过程的影响(图1C、1D)。随着退火的进行,两个样品的峰位于约14.4°处的高角度发生明显的变化,表明由于DMA蒸发引起的晶格收缩导致了中间相(CsxDMA1−x)PbI3的分解最终完全消失。在0分钟的初始阶段,两种铸造的钙钛矿薄膜显示出相似的衍射峰,这些峰被称为DMAPbI3相。随着退火的进行,DMAPbI3相开始分解,γ-CsPbI3相逐渐形成。更仔细地观察位于约11.8°处的DMAPbI3峰的衍射强度演变(图1E),衍射强度在短时间内随着退火时间的增加而改善,但在较长的退火时间下急剧下降,对于对照样品和PAA样品都是如此。对于位于约14.4°处的CsPbI3的(110)峰,两个样品的衍射强度逐渐增加,表明CsPbI3的形成。这一现象表明,PAA处理后,包括对照和PAA样品在内的两者都经历了三个阶段,即(1)DMAPbI3的结晶,(2)DMAPbI3的分解,和(3)γ-CsPbI3的形成。有趣的是,经过PAA处理后,DMAPbI3的分解和高结晶度γ-CsPbI3的相变过程加速进行。
进行了原位的斜入射宽角X射线衍射(GIWAXS)测量,提取并绘制了DMAPbI3(100)峰和CsPbI3(110)峰的强度演变如图1F所示。对于对照样品,在390秒的退火后,DMAPbI3开始分解,表现为DMAPbI3(100)峰的强度下降。通过PAA的引入,DMAPbI3的分解在310秒开始,这表明PAA有效地提前了DMAPbI3的分解。就CsPbI3的演变来看,对于对照样品和PAA样品,CsPbI3的形成在310秒开始。没有PAA的引入,CsPbI3(110)峰的强度持续增加,直到1,240秒,导致了相对较长的晶化周期,达到840秒。相反,PAA样品在明显减少的540秒内完全结晶。PAA样品在CsPbI3结晶期间的较陡坡度表明PAA加速了CsPbI3的形成动力学(图1F)。DMAPbI3辅助CsPbI3结晶过程是由DMAPbI3的分解动力学(即DMAI升华)和CsPbI3的形成动力学(即Cs离子插入Pb-I框架)共同决定的。DMAPbI3分解与CsPbI3形成的不匹配可能导致在对照样品中观察到的CsPbI3薄膜中出现小孔洞(图1G、图S8和图S9),通常这些小孔洞会作为载流子复合位点,妨碍PCE的提高。
要点2:氢键对DMA提取的促进
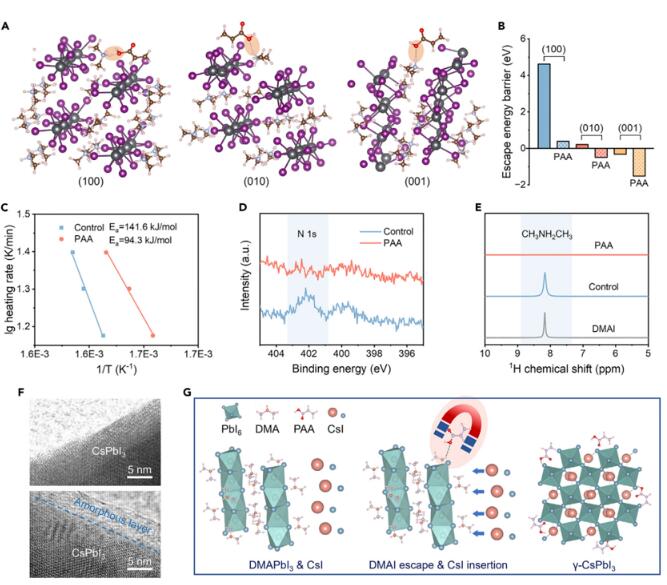
为了从热力学角度深入了解PAA对CsPbI3结晶的影响,研究了PAA添加剂对CH3NH2CH3+(DMA+)从DMAPbI3中逃逸性质的影响,通过密度泛函理论(DFT)进行了研究。建立了DMAPbI3模型的(100)、(010)和(001)表面(图2A和图S10)以计算在有无PAA处理情况下的逃逸势垒(Eb)。显然,所有经过PAA处理的三个表面的Eb值明显低于对照DMAPbI3的Eb值(图2B;表S1),这归因于PAA与DMAPbI3之间的强氢键作用(N–H···O,用橙色标示)。进一步对经过PAA处理或未经处理的样品进行了热重分析(TG)测量,以定量评估氢键对DMAPbI3的热分解的影响。将1%质量损失位置的温度定义为分解温度。与不同加热速率下的对照样品相比,PAA样品在各种加热速率下明显降低了分解温度。通过Ozawa-Flynn-Wall方法计算了两个样品热分解的活化能(Ea)。PAA样品的Ea明显低于对照样品的141.6 kJ/mol,为94.3 kJ/mol(图2C),这验证了理论结果。
图2 氢键促进二甲基铵萃取机理
进行X射线光电子能谱(XPS)光谱测量,以检测形成的钙钛矿表面的成分变化。对照样品显示位于约402 eV处的峰(图2D),为残留的DMA阳离子的N 1s。对于PAA样品,N 1s峰消失,这意味着在钙钛矿薄膜表面没有残留的DMA。然后,通过将制备的CsPbI3薄膜溶解在DMSO-d6溶液中,利用核磁共振(NMR)测量来检测整个薄膜中残留的DMA。位于8.17 ppm处的峰是DMA分子。对于对照样品,DMA信号仍然存在(图2E)。对于PAA样品,DMA信号消失。透射电子显微镜(TEM)证实了CsPbI3薄膜中存在PAA。与对照样品相比(图2F),经过PAA处理的CsPbI3样品被覆盖了几纳米的非晶层,这是因为大尺寸的PAA无法进入晶格并在晶粒边界之间聚集。非晶层的形成也可能有助于提高器件稳定性。根据这些分析,图2G中描述了一个三阶段的结晶过程:(1)湿膜最初由DMAPbI3、PAA和CsI组成,然后在退火过程中,由于PAA引起的氢键有助于DMA的提取,并加速了CsI插入PbI6骨架,从而快速转变为CsPbI3。(2)延长的退火导致DMA的完全去除,同时PAA位于晶粒边界,形成γ-CsPbI3。(3)氢键促进的DMA提取概念通过使用两种类似分子的聚丙烯腈(PAN)和聚(4-乙烯吡啶)(PVP)进一步得到验证,这些分子可以与DMAPbI3形成N–H···N氢键相互作用。如预期,PAN和PVP都可以加速CsPbI3的结晶并抑制DMAPbI3残留,证明了该方法的普遍性。
要点3:薄膜光与器件的的表征
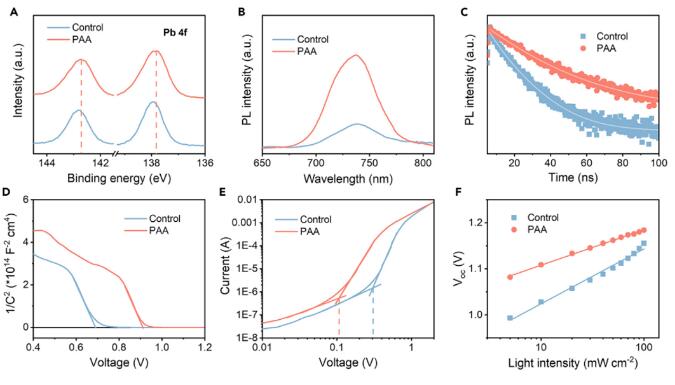
PAA样品的Pb 4f峰相对于对照样品显示出明显的向较低的束缚能量的移位(图3A)。束缚能量的移位表明了一种有利的钝化效应,这可能是由PAA中的羰基引起的。在对照和PAA样品上进行了稳态和时间分辨的光致发光(PL)测量。经PAA处理的CsPbI3薄膜的PL发射强度比对照薄膜高3倍(图3B)。通过拟合PL衰减曲线,PAA-CsPbI3薄膜的平均寿命(τ-average)为29.12 ns,明显长于对照样品的13.29 ns表明抑制了缺陷引起的非辐射复合损失。对于空间电荷有限电流(SCLC)测量,经过PAA处理,CsPbI3样品的缺陷密度明显降低(4.02×1014 cm-3),相对于对照样品(1.17×1015 cm-3)(图3D)。PAA处理的CsPbI3 PSC具有更高的内建电位(Vbi,0.91 V),而对照样品为0.69 V(图3E),这归因于PL和SCLC结果所示的抑制缺陷密度。增加的Vbi有助于形成更长的耗尽区域和更强的驱动力用于载流子的传输和分离。进一步进行了光强度依赖的J-V测量,以研究Voc和光强度之间的关系。在300 K下,PAA-CsPbI3器件的斜率减小为1.28 kT/q,而对照样品为1.73 kT/q,这是由于膜质量的改善和缺陷密度的降低所致(图3F)。
图3光学和器件电学特性
要点4:PSCs的PV性能和稳定性
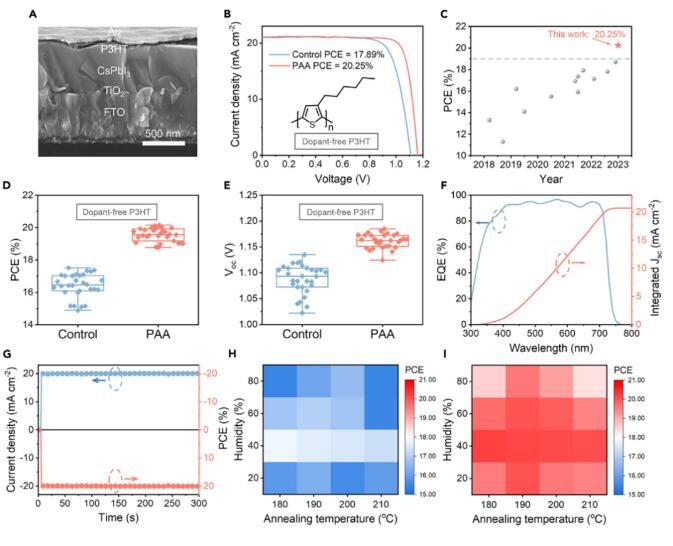
作者制备了无机PSCs以评估PAA处理对器件光伏性能的影响。为避免由含水性添加剂引起的器件降解,选择不含杂质的聚合物P3HT(图4A)作为空穴传输层。有趣的是,PAA处理的CsPbI3 PSCs取得了20.25%的PCE,Voc为1.16 V,Jsc为20.99 mA cm-2,FF为0.831(图4B),这是报道的无掺杂HTL的n-i-p结构无机CsPbI3PSCs中最高的PCE(图4C;表S3)。平均效率也从16.42%显著提高至19.55%(图4D和4E;表S4),主要是由于Voc(从1.08提高到1.16 V)和FF(从0.73提高到0.81)的增加,这是由于抑制了DMAPbI3残留引起的缺陷复合。从外部量子效率(EQE)谱中积分得到的电流密度(图4F)为20.7 mA cm-2,与J-V曲线获得的Jsc值非常匹配(偏差不超过2%)。冠军器件提供了20.08%的稳态PCE(图4G)和小的滞后。借助目前的氢键促进的DMA提取方法,可以在惊人宽广的加工窗口下制备高性能的CsPbI3PSCs(PCE超过20%),例如广泛的退火温度范围从180°C到210°C和广泛的湿度范围从20%到60%相对湿度(图4H和4I)。
图4 无掺杂HTLs的CsPbI3 PSCs的光伏性能
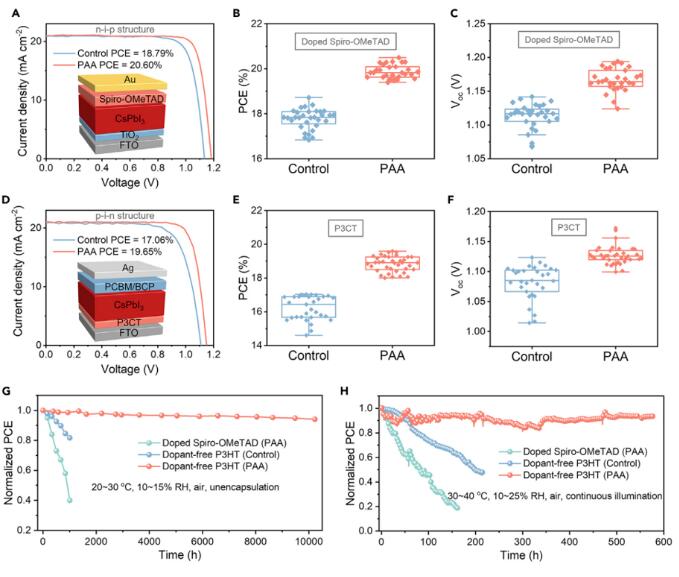
为探索目前的氢键促进的DMA提取方法的普适性和兼容性,对其他两种常用的HTL和各种器件结构进行了研究,即spiro-OMeTAD用于n-i-p结构和聚[3-(4-羧基丁基)噻吩-2,5-二基](P3CT)用于p-i-n结构。对于n-i-p结构的PSCs,使用spiro-OMeTAD HTL,经过PAA处理的器件实现了20.60%的高PCE,Voc为1.18 V,Jsc为20.95 mA cm-2,FF为0.834(图5A)。与基于P3HT的PSCs类似,经PAA处理的PSCs表现出明显的平均PCE增加(图5B),从17.77%提高到19.85%,这是由于Voc从1.11提高到1.17 V(图5C)。此外,PAA处理的器件表现出小的滞后现象(图S23)。对于使用P3CT HTL的p-i-n结构PSCs,经PAA处理的PSCs也表现出改善的器件性能,达到了19.65%的高PCE(图5D),并具有小的滞后现象(图S24)。倒置PSCs显示出明显的平均PCE增加(从16.23%提高到18.83%;图5E)和Voc的增加(从1.08提高到1.13 V;图5F)。
图5 不同器件结构和HTLs的CsPbI3 PSC的性能和稳定性
经PAA处理的掺杂spiro-OMeTAD HTL的未封装PSC的PCE在储存在湿度(<15%相对湿度)环境中经过1,000小时后急剧下降到初始PCE的40%(图5G和图S25)。而未经PAA处理的掺杂spiro-OMeTAD HTL的对照PSC在1,000小时后下降到初始PCE的81.7%。相比之下,经PAA处理的无掺杂P3HT HTL的PSC在老化10,224小时后保持约94%的初始效率,表现出卓越的稳定性。从操作稳定性的角度看,经PAA处理的带有掺杂spiro-OMeTAD HTL的PSC在连续照明161小时后PCE迅速下降到初始PCE的约20%(图5H和图S26)。具有无掺杂P3HT HTL的对照器件在照明214小时后下降到初始PCE的约48%。相比之下,经PAA处理的无掺杂P3HT HTL的PSC在连续照明576小时后保持了93%的初始效率。这种稳定性的提高可能与通过在PAA和CsPbI3之间形成氢键(O-H···I)抑制碘空位有关。这些结果表面,氢键促进的DMA提取方法结合使用无掺杂P3HT HTL显著改善了PSCs的稳定性,因为它提高了CsPbI3薄膜质量,并消除了吸湿性掺杂剂。
四、小结
作者开发了一种氢键促进的DMA提取方法,用于制备高质量的γ-CsPbI3薄膜。PAA和DMAPbI3之间的氢键(N-H···O)减小了DMA的逃逸能,加速了CsPbI3的结晶动力学,导致了DMAPbI3残留物的完全消除和无针孔的γ-CsPbI3薄膜的形成。通过使用类似的PAN和PVP分子验证了这一概念,它们也可以与DMAPbI3形成N-H···N氢键。此外,PAA和CsPbI3之间的氢键(O-H···I)抑制了碘空位,使得晶化过程更加环保,并显著提高了器件的稳定性。在宽窗口的湿度(20%∼60% RH)和退火温度(180°C∼210°C)条件下,具有无掺杂P3HT HTLs的PSCs可以获得超过20%的PCE(冠军PCE为20.25%)。在低湿度条件(<15% RH)下老化10,224小时后,PSCs保持了94%的初始PCE,而在连续照明570小时后仍然保持了93%以上的PCE。这一策略通过氢键工程为制备高效稳定的无机PSCs开辟了新的途径。
五、参考文献
Ming-Hua Li, Shuo Wang et al. Hydrogen-bonding-facilitated dimethylammonium extraction for stable and efficient CsPbI3 solar cells with environmentally benign processing, Joule
Doi: 10.1016/j.joule.2023.09.009 (2023).
 邮箱: info@chemborun.com
邮箱: info@chemborun.com 电话: +86-574-87178138
电话: +86-574-87178138  中国浙江省宁波市江南路1558号(大陆)/315010
中国浙江省宁波市江南路1558号(大陆)/315010