Conducting CO2RR in acidic electrolytes can improve CO2 utilization by reducing bicarbonate formation and CO2 crossover. However, the selectivity of electrochemical reduction of CO2 is reduced by the competitive hydrogen evolution reaction, where the formation of CO and its subsequent coupling are crucial to achieve the formation of multicarbon (C2+) products. The two reactions rely on different catalyst properties that are difficult to achieve in a single catalyst.
With this in mind, Edward H. Sargent et al. of the University of Toronto achieved the desired conversion by running two different catalyst layers in series, decoupling the CO2-to-C2+ reaction into CO2-to-CO and CO-to-C2+ Two steps. The first layer of catalyst is atomically dispersed cobalt phthalocyanine, which can reduce CO2 to CO with high selectivity. This process increases local CO availability to enhance the C-C coupling step on the second catalyst layer. The second catalyst layer is a Cu nanocatalyst with a Cu-ionomer interface. The optimized series electrode can achieve 61% C2H4 Faradaic efficiency and 82% C2+ Faradaic efficiency at 25°C and 800mAcm−2 current. When optimized for single-pass utilization, the system achieved a single-pass carbon efficiency of 90±3% at 800mA cm−2 at a CO2 flow rate of 2 ml min−1, while the C2H4 Faradaic efficiency was 55±3%, and the total C2+ Faraday efficiency is 76±2%.
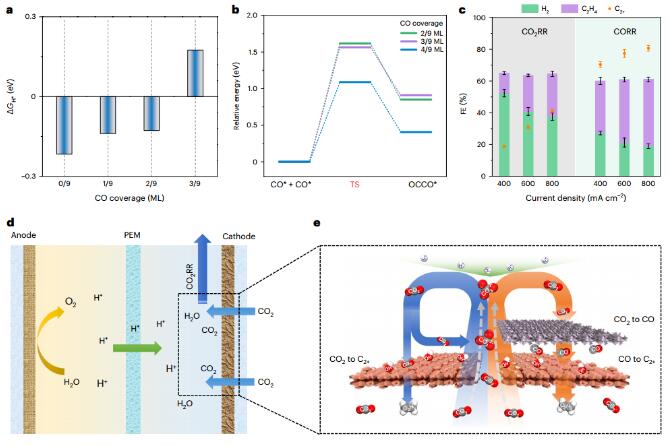
CO2RR spatial decoupling strategy
The authors developed strategies to promote selective C2+ formation under acidic conditions. Using DFT calculations, the authors found that greater CO coverage on Cu reduces the Gibbs free energy of H adsorption (ΔGH*), resulting in a HER overpotential Increase. It was also found that the higher the CO coverage on Cu, the lower the energy barrier of the C-C coupling reaction. Experiments have also confirmed that increasing CO coverage can increase C2H4 and C2+ production. Therefore, in order to improve C2+ selectivity, it is necessary to develop ways to generate local high concentrations of CO on the catalyst surface. The authors adopted a spatial decoupling strategy to connect the CO2 to CO conversion catalyst and the CO to C2+ conversion catalyst in series.
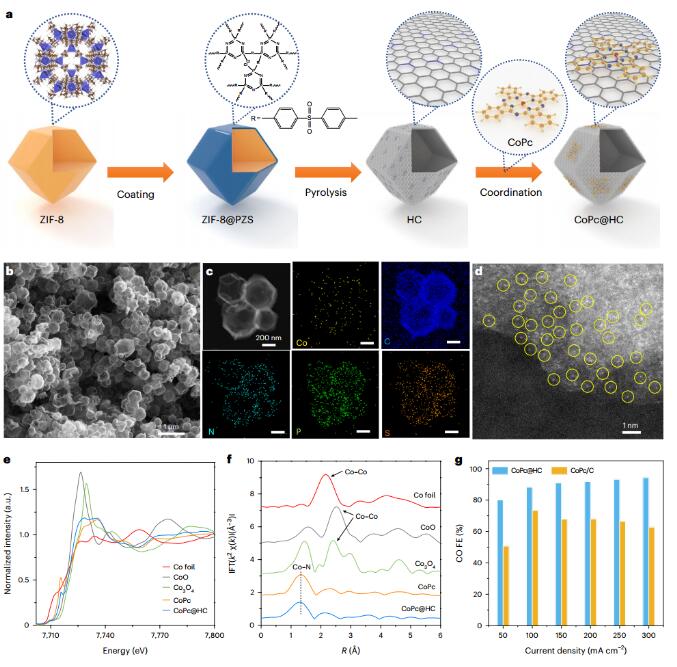
Synthesis and characterization of atomically dispersed CoPc@HC
The authors chose cobalt phthalocyanine to catalyze the conversion of CO2 into CO, and prepared hollow carbon (HC)-supported CoPc (CoPc@HC) through thermal depolymerization, in which a single CoPc molecule was anchored on the scaffold. The CoPc@HC catalyst has a hollow morphology and no CoPc agglomeration. High-resolution Z-contrast images confirm the atomic dispersion of Co, and X-ray absorption spectroscopy demonstrates strong catalyst-support interactions, which reduce Co agglomeration. The results show that there are electronic interactions between CoPc and N species on the HC support. BET measurements show that CoPc@HC has a surface area of 1,087 m2g−1, which may contribute to the mass transport of CO2 gas and intermediates. The CoPc@HC catalyst has 94% CO FE and good stability. Next, the authors considered combining the CO2-CO and C-C coupling steps on a single support.
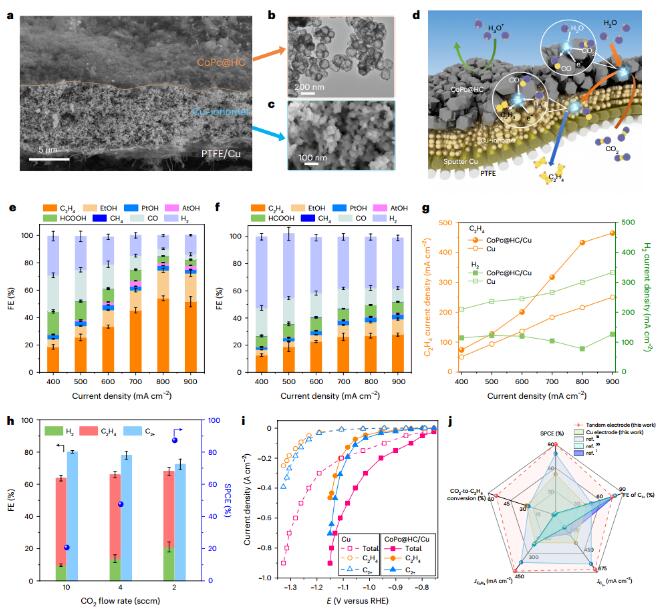
CO2RR performance of series catalysts
The authors designed a series electrode, consisting of a C-C coupling catalyst and an upper CO2-to-CO catalyst, by increasing the density of copper sites and enhancing the mass transfer of carbon dioxide to these active sites, between CoPc@HC and sputtered Cu. A three-dimensional Cu-ionomer interface catalyst layer composed of ionomer-coated Cu nanoparticles (NPs) was introduced to construct a CoPc@HC/Cu series electrode. By adjusting the size of Cu NPs and the type of ionomer and changing the electrode configuration to optimize the CoPc@HC/Cu tandem electrode, the FE of C2H4 increased from 30% to 54% and the C2+ increased from 36% to 80%. Compared with Cu electrodes, CoPc@HC/Cu tandem electrodes exhibit higher C2H4 and C2+ current densities at lower potentials. A full cell voltage of 3.8V was achieved at 500mAcm−2, resulting in a C2H4 energy efficiency of 16%.
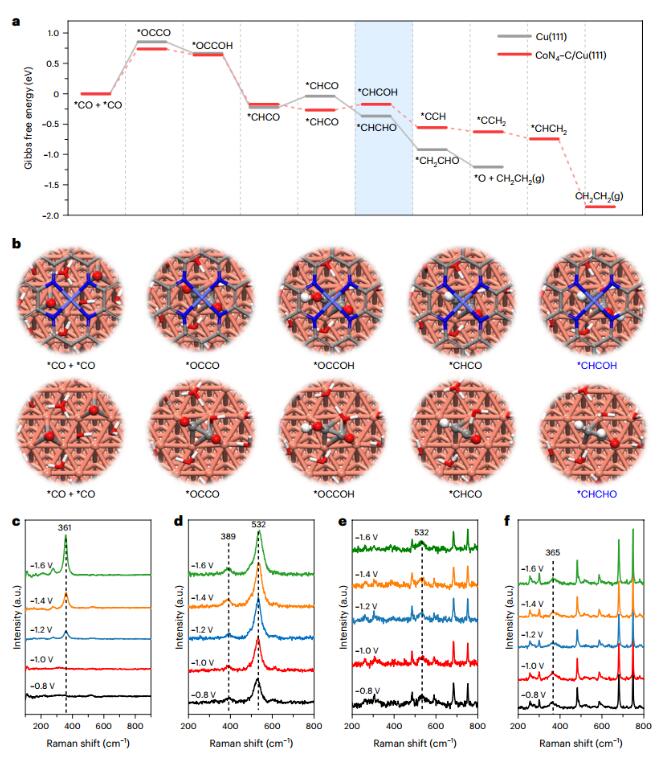
Mechanism analysis
The authors further studied the interface structure between CoPc@HC and Cu nanoparticles. High-resolution TEM images and energy dispersive spectrograms and intensity distributions indicate the existence of sub-nanometer spacing between CoPc@HC and Cu NPs. The CO2RR pathway of periodic Cu plates covered with CoN4-C layers was studied by DFT, and the results showed that the combination of CoPc and Cu affected C2H4 selectivity in addition to promoting C-C coupling. The authors further increased the spacing between CoPc@HC and Cu particles by constructing a CoPc@HC/(Cu+CoPc@HC) series electrode, with a C2H4 FE of 61% and a C2+ FE of 82% at 800mAcm–2. To elucidate the interaction between CO and the tandem electrodes, we probed CO adsorption on the Cu surface using in situ Raman spectroscopy. The results show that uniform coverage of CoPc molecules on Cu is necessary for the tandem catalysis concept, a finding that promotes the application of atomically dispersed CoPc@HC.
References
Chen, Y., Li, XY., Chen, Z. et al. Efficient multicarbon formation in acidic CO2 reduction via tandem electrocatalysis. Nat. Nanotechnol. (2023).
 E-mail: info@chemborun.com
E-mail: info@chemborun.com Tel: +86-574-87178138
Tel: +86-574-87178138  No. 1558, Jiangnan Road,, Ningbo, Zhejiang, China (Mainland)/31
No. 1558, Jiangnan Road,, Ningbo, Zhejiang, China (Mainland)/31