1. The addition of lead chelating molecules in HTLs can strongly interact with lead (II) ions (Pb2+), resulting in a reduction of amorphous regions near the HTLs and passivation of the underlying surface of the perovskite.
2. The minimum module with an aperture area of 26.9 square centimeters achieves a power conversion efficiency (PCE) of 21.8% (stable at 21.1%), corresponding to a minimum cell efficiency of 24.6% (stable at 24.1%) over the entire module area.
3. Small-area cells and large-area minimum modules containing lead chelating molecules exhibit photostability for 3010 hours and 2130 hours, respectively, with an efficiency loss of 10% compared to the initial value under daily illumination and open-circuit voltage conditions.
一、Attention to the interface between perovskite and hole transport layers (HTLs) in small-area p-i-n structured perovskite solar cells
The power conversion efficiency (PCE) of small-area n-i-p structured single-junction perovskite solar cells has reached the level of single-crystal silicon solar cells (>25%). However, the PCE of p-i-n structured perovskite solar cells is lower, and there is significant loss after transferring small cells to modules, mainly due to non-uniformities in the perovskite or charge transport layers. The certified PCE of perovskite-based modules is still low, approximately 19%. Several strategies have been adopted to further improve the efficiency of p-i-n structured devices, such as improving crystallization processes and passivating surface defects. For example, when surface treatment is used to enhance charge extraction, the certified stable PCE of small-area p-i-n structured devices can exceed 24%. However, compared to the top surface, less attention has been paid to the perovskite-hole transport layer (HTL) interface. Nonradiative recombination of interface charge carriers caused by buried interfacial defects may limit the efficiency of p-i-n structured perovskite solar cells. Photoluminescence (PL) quantum yields of perovskite near the HTL have been observed to be significantly lower.
二、Summary of achievements
In light of this, a team led by Jinsong Huang from the University of North Carolina at Chapel Hill in the United States reported an effective method to reduce the amorphous regions at the bottom of perovskite thin films. The method involved embedding lead chelating molecules (LCMs), including a widely used electron transport blocking material, BCP, into HTLs. BCP, along with DMSO, strongly interacts with lead ions through complexation, reducing the residual amount of DMSO and thus reducing the amorphous regions in the perovskite near the HTL. BCP also passivates the perovskite, improving the PCE, repeatability, and stability of perovskite solar cells and modules.
三、Results and discussion
Key Point 1: Strong interaction between perovskite and LCMs
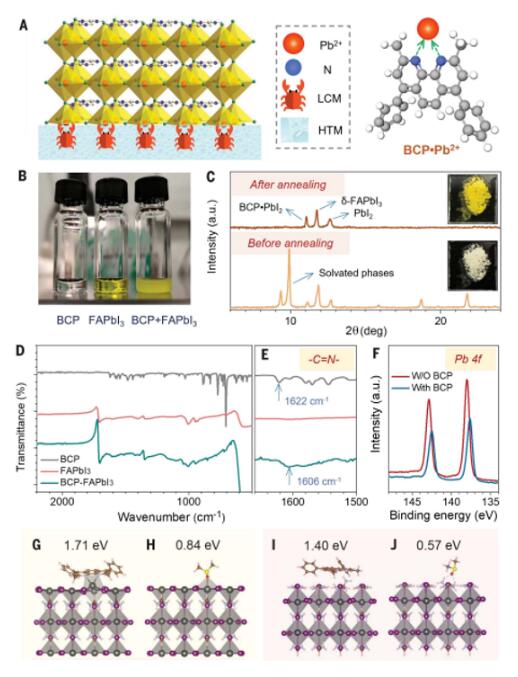
We passivate the bottom perovskite-HTL interface by mixing a passivating agent into the HTL. We use LCMs, which are used for water purification or as drugs for lead poisoning treatment, as these molecules should strongly interact with Pb2+ in the perovskite at the interface with the HTL (Figure 1A). The molecular structures of selected LCMs, S1 and S2, including BCP, TSA, L-ascorbic acid (VC), meso-2,3-dimercaptosuccinic acid (MDSA), and ethylenediaminetetraacetic acid (EDTA), provide two or more lone-pair electrons for chelation with Pb2+ on the perovskite surface. The chelation of LCMs with Pb2+ leads to the precipitation of Pb2+ after being added to the perovskite precursor solution. We chose BCP for demonstration as its solubility in 2-ME is low, less than 0.2 mg mL−1, which is lower than the rapid blade-coating process in our perovskite inks. When BCP is mixed with a solution of formamidinium lead iodide (FAPbI3), even at a very low BCP concentration of <0.05 mg mL−1, a pale yellow precipitate forms (Figure 1B), indicating that the solubility of the reaction product is even lower than that of BCP. The X-ray diffraction (XRD) pattern of the pale yellow precipitate (Figure 1C and Figure S4) reveals diffraction peaks of multiple compounds, including PbI2•BCP and some solvated phases. The peak at 11.06° is assigned to PbI2•BCP, as it is observed in the precipitate from the mixture of PbI2 and BCP solutions (Figure S5). However, all solvated phases, such as PbI2•DMSO, disappear upon annealing the precipitate to remove DMSO at 100°C, and the powder color changes from yellow to pale yellow. The presence of PbI2•BCP in the annealed powder indicates a stronger interaction of BCP with Pb2+ than DMSO with Pb2+. Fourier-transform infrared spectroscopy (FTIR) measurements were conducted to study how BCP interacts with the perovskite precursor (Figure 1D and 1E). The C=N stretching of BCP at 1622 cm-1 can be easily distinguished from the stretching at 1720 cm-1 in FAPbI3. After mixing with FAPbI3, the C=N stretching peak of BCP shifts to a lower wavenumber at ~1606 cm−1, consistent with the movement of electron density toward Pb2+, indicating a strong interaction between BCP and Pb2+. The same FTIR shifts are observed when all other LCMs are mixed with FAPbI3 (Figure S6), supporting the Pb2+ chelation effect of these molecules. BCP has two pyridine rings in its structure. We note that the C=N FTIR peak is shifted completely instead of splitting into two peaks. This suggests that the N atoms in both pyridine groups of BCP interact with Pb2+, resulting in chelation of BCP with Pb2+. X-ray photoelectron spectroscopy (XPS) also confirms the interaction of BCP with Pb2+, as the Pb 4f peak shifts upon covering the perovskite film with a thin layer of spin-coated BCP (Figure 1F). The decrease in binding energy of Pb2+ is consistent with the chelation of BCP with Pb2+ due to electron sharing between N (in BCP) and Pb2+.
The chelation effect provides a stronger interaction with the perovskite compared to a single pyridine group. We calculated the binding energies of BCP and DMSO molecules on the (100) FAPbI3 perovskite surface with PbI2 termination and formamidinium iodide (FAI) termination. During the PbI2 termination, the two N atoms of BCP bond with a single Pb atom of FAPbI3 (Figure 1G), while for DMSO, only an O-Pb bond is formed (Figure 1H). The binding energy of BCP with FAPbI3 is 1.71 eV, compared to 0.84 eV for DMSO. For the FAI termination, hydrogen bonding occurs between the two N atoms of BCP and one H atom of FA (Figure 1I). The binding energy of BCP with FAPbI3 is 1.41 eV. For DMSO, hydrogen bonding occurs between the O atom of DMSO and two H atoms of FA+ (Figure 1J), resulting in a smaller binding energy of 0.57 eV. On both surfaces with PbI2 and FAI terminations, the binding energy of BCP is two to three times higher than that of DMSO.
Figure 1: Chelation of lead ions by BCP
Key Point 2: Defect Passivation of the Bottom Interface by LCMs
We introduced LCMs into the poly [bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) layer by mixing them in different proportions in toluene for BCP and TSA, or by spin-coating them onto the PTAA layer to obtain insoluble LCMs, including VC, MDSA, and EDTA (see Supplementary Methods for details). The perovskite composition of FA0.9Cs0.1PbI3 was used for device fabrication, as it has been reported to exhibit high operational stability. Both the HTL and perovskite layers were coated under ambient conditions using a nitrogen-assisted blade for deposition. We performed steady-state photoluminescence (PL) and PL lifetime measurements to investigate how the LCMs embedded in PTAA affect the perovskite-HTL interface. Previous studies have suggested that the embedded perovskite-HTL interface may be more defective than the top surface of the perovskite, depending on the processing conditions.
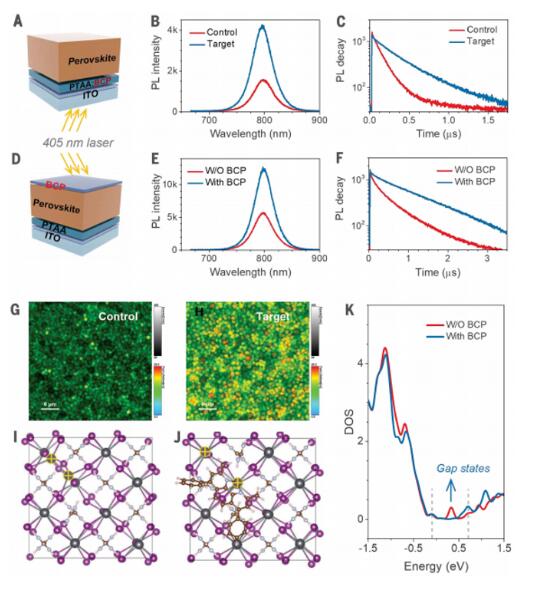
When we sandwiched the PTAA-coated perovskite thin film between two identical charge-extraction conditions and compared the PL intensity at the top surface with that at the bottom surface, we also observed a approximately 40% decrease in PL at the bottom surface (Figure S7), indicating indeed a higher defect density at the bottom interface. When we probed the bottom interface by introducing the excitation light from the indium tin oxide (ITO) side (Figure 2A), BCP increased the steady-state PL intensity by a factor of 1.7 (Figure 2B). The PL lifetime at the bottom of the perovskite film also increased from 195 ns to 452 ns (Figure 2C), suggesting that pre-embedding BCP in PTAA reduced the nonradiative recombination defects at the bottom perovskite interface. However, the prolonged PL lifetime could also be attributed to the generation of a more crystalline perovskite layer by the HTL. To validate the passivation function of BCP, we spun a BCP layer on top of the perovskite to exclude the influence of perovskite crystallinity. We measured the changes in PL spectra and PL lifetime when the light was incident from the air side (Figure 2D). BCP increased the PL intensity by a factor of 1.2 and extended the PL lifetime to 1.43 ms (Figure 2E and F), confirming the passivation effect of BCP on the perovskite and indicating that the addition of BCP also modified the morphology of the perovskite close to this interface.
We evaluated the uniformity of passivated perovskite-HTL interfaces using fluorescence lifetime imaging microscopy (FLIM). Subsequently, we referred to pure PTAA and BCP:PTAA HTLs as control and target, respectively. Grains were discerned in the PL maps and exhibited an apparent size of 1 to 2 millimeters (Figure 2G and H). Within a measurement area of 40 mm x 40 mm, the PL intensity and lifetime were relatively uniform for both the control and target films, but the target film exhibited a 1.5-fold increase in PL intensity and a 1.1-fold increase in PL lifetime compared to the control film. The passivation effects of BCP and DMSO were further compared by DFT calculations. We found that perfect PbI2- and FAI-terminated surfaces do not form deep-gap states.
Pb-Pb dimers can form additional I vacancies on the surface, which can be easily formed by the loss of iodides. Pb-Pb dimers induce gap states through Pb 5p–Pb 5p bonding. As revealed by previous studies on the (100) surface terminated with PbI2, BCP can eliminate deep-gap states, whereas DMSO cannot. Pb-Pb dimers formed by the presence of two iodide vacancies generate deep-gap states (Figure 2I). When BCP molecules interact with Pb-Pb dimers, both N atoms bond with one Pb atom of the dimer, disrupting the dimer structure (Figure 2J) as the Pb-N bonds partially saturate Pb coordination. As shown in Figure 2K, the disappearance of the gap states occurs after the rupture of the Pb-Pb dimer. However, DMSO cannot break the Pb-Pb dimer as it only forms a Pb-O bond. The Pb-Pb dimer is retained after DMSO molecules bind, leaving behind the gap states (Figure S8, A and B).
Figure 2: Function of BCP in reducing perovskite defects
Key Point 3: Reduction of Amorphous Phases in Perovskite by LCMs
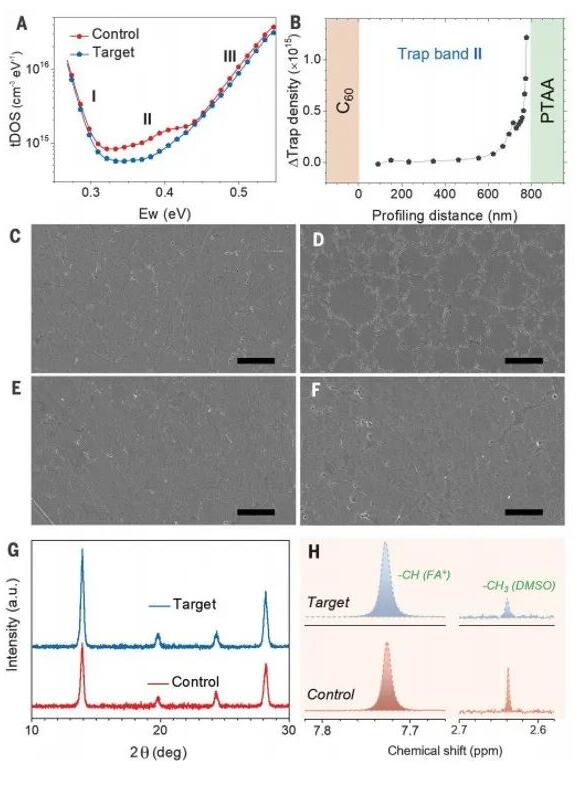
To identify the defects reduced by introducing BCP into PTAA, we measured the trap density changes of the devices using thermal admittance spectroscopy and drive-level capacitance profiling (DLCP). The trap density of states (tDOS) curves in Figure 3A show three distinct trap bands for both the control and target devices, similar to those reported for iodide perovskites. These trap bands are assigned to negatively charged iodine vacancies (Ii-) and positively charged iodine vacancies (Ii+). The density of Ii- does not show significant changes in the perovskite, while the density of Ii+ is reduced by introducing BCP into PTAA. DLCP measurements provide consistent results, as the density of Ii- remains unchanged throughout the device, while the density of Ii+ is significantly reduced (Figure S9).
We plotted the change in Ii+ density (△Ii+ = Ii+,target - Ii+,control) in the perovskite from C60 to the HTL side induced by BCP (Figure 3B). It is evident that BCP primarily reduces the density of Ii+ in the perovskite near the HTL. Our recent study on another perovskite with the same composition indicated a similar reduction in Ii+ density during thin-film thermal annealing, while the density of Ii- remained unchanged for hundreds of hours in the dark. Since the decrease in Ii+ density is accompanied by a reduction in amorphous phases and the appearance of related vacancies near the bottom interface close to PTAA, we hypothesize that the amorphous phase at the bottom of the perovskite film may be rich in Ii+ and is reduced by the BCP:PTAA HTL.
To validate this hypothesis, we performed scanning electron microscopy (SEM) measurements to examine the voids caused by recrystallization of the amorphous regions in the control perovskite devices using PTAA as the HTL. These devices were encapsulated to avoid the influence of moisture and oxygen and were soaked for approximately 1000 hours under indoor lighting to allow for recrystallization to occur. The perovskite films were peeled off from the ITO/PTAA substrates, exposing the bottom surface, and then subjected to SEM analysis. As shown in Figure 3C to F, the average grain size of these perovskite films ranged from 1 to 2 mm, consistent with the PL mapping results. There were no apparent morphological differences at the bottom of the perovskite films for both fresh samples with and without BCP. After aging, numerous shallow voids appeared around the grain boundaries in the aged control sample, similar to the results of thin-film thermal annealing. In contrast, the bottom morphology of the aged target sample showed little difference from the fresh target sample, indicating that BCP significantly reduces the formation of amorphous phases at the bottom of the perovskite film. Grazing incidence X-ray diffraction (GIXRD) measurements were performed in the mode of the fresh samples directly near the perovskite crystals close to the HTL. As shown in the GIXRD patterns in Figure 3G and Figure S10, the peak of the (100) orientation not only increased in intensity by 60% but also sharpened by 8%, confirming the improved crystallinity of the perovskite bottom layer induced by BCP. The reduction in amorphous phases is expected to contribute to the decrease in Ii+ density at the bottom of the perovskite film (Figure 3A).
We investigated why BCP can reduce the amorphous phases. Previous studies have indicated that captured solvent DMSO can also lead to the formation of amorphous phases at the bottom and grain boundaries of the perovskite. We measured the amount of captured DMSO in the perovskite film after thermal annealing using proton nuclear magnetic resonance (1H-NMR) spectroscopy. The perovskite films manufactured on different HTLs were scraped off the substrates and dissolved in deuterated oxide. The content of DMSO in the perovskite film was determined by the ratio of the characteristic peaks of DMSO at 7.71 ppm and 2.63 ppm to the characteristic peak of FA+. As shown in Figure 3H and Figure S11, the DMSO/FA+ atomic ratio in the control sample was 0.13%, while it was 0.08% in the target sample (Table S1).
Therefore, BCP in PTAA indeed reduces the amount of captured DMSO, as BCP competes with DMSO for coordination with Pb2+ in the bottom region during film formation, thus reducing the amount of DMSO bound to Pb2+ (Figure 1C). The reduction in captured DMSO leads to a decrease in the amorphous degree of the perovskite. BCP reduces defective amorphous perovskite, thereby improving device efficiency and stability. The capture of DMSO may be highly uneven, especially for large-area perovskite films coated on the production line, depending on the local vapor pressure, which can vary depending on the location. The reduced capture of DMSO explains the improved uniformity of the perovskite film and holds promise for enhancing the efficiency of perovskite modules.
Figure 3: Influence of BCP on perovskite crystallization
Key Point 4: Enhancing the Performance of Perovskite Solar Cells with LCMs
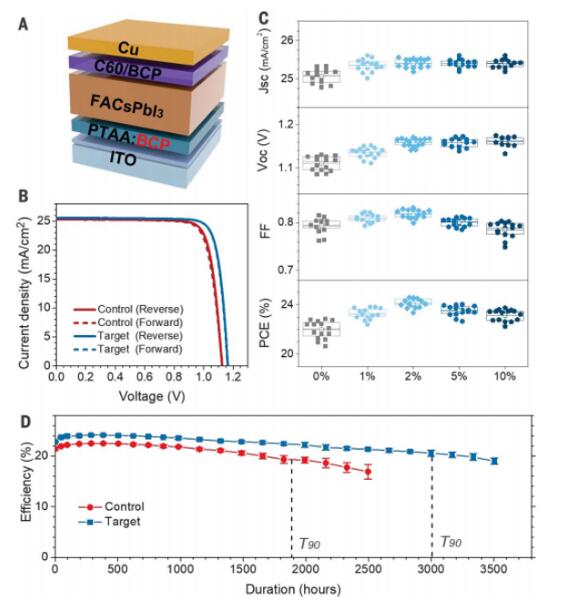
To assess how LCMs affect the performance of perovskite solar cells, perovskite solar cells were fabricated with an ITO/HTL/perovskite/C60/BCP/Copper (Cu) structure (Figure 4A). As shown in Figure S12, the integration of LCMs into or on top of the HTL improved the stability of all devices during accelerated light-induced degradation testing (200 mW cm-2, open-circuit and ambient conditions, 59°C to 65°C). Among all the tested LCMs, BCP and TSA resulted in higher power conversion efficiency (PCE) and improvements in open-circuit voltage (VOC) and fill factor (FF) (Figure S13). The higher PCE can be attributed to better blending of BCP or TSA with PTAA, as the phenyl groups in BCP or TSA are expected to induce π-π interactions with PTAA, facilitating the transfer of charge carriers from the perovskite to PTAA. Another reason is the lower solubility of BCP and TSA in perovskite solvents. We chose BCP for PCE optimization as BCP has been widely used as an electron transport material in p-i-n structured perovskite solar cells and its hole-blocking properties have been frequently reported.
Thus, it is necessary to optimize its loading in PTAA to achieve optimal hole extraction. Perovskites have a sufficiently long carrier diffusion length, so even if some local contacts are blocked, the photogenerated charges can still be effectively extracted, as demonstrated in nanocontact perovskite solar cells or solar cells with discontinuous insulating interface layers. The short-circuit current density (JSC) of the control device, with a working area of 0.08 cm2, was 25.2 mA cm−2, VOC was 1.13 V, FF was 0.80, and PCE was 22.7% (Figure 4B), consistent with our previous results. As shown in Figure 4C, approximately 2 wt% of BCP in PTAA was required to increase the FF of the perovskite device. When the BCP loading in PTAA exceeded 2 wt%, the device FF decreased, possibly due to reduced hole collection. The same trend of change in JSC and FF was observed at different BCP loading rates.
In contrast, the VOC of the device increased from 1.13 V to 1.17 V as the BCP loading rate increased from 0 to 2 wt%. The VOC saturated with further increases in the BCP ratio, as hole extraction is not required under open-circuit conditions. The champion target device with optimal BCP exhibited JSC of 25.5 mA cm-2, VOC of 1.17 V, FF of 0.825, and PCE of 24.6% (Figure 4B). The average PCE increased to 24.1%, and the hysteresis phenomenon was not significant in both types of devices. The observed slight variation in the PCE of small-area devices (0.08 cm2) is consistent with the uniform distribution of BCP in PTAA and uniform passivation. The average PCE of the control group was approximately 22.0%, which is consistent with results reported using perovskite compositions containing CsI or FAI. Considering that BCP has other functional groups besides the phenylpyridine, we evaluated two additional molecular groups with these functionalities and confirmed the dominant improvement in device efficiency due to chelating phenylpyridine groups (Figure S14 and S15).
Pre-embedded BCP in PTAA also significantly improved the stability of the devices. Stability measurements were performed using three different light sources based on device area and light source lifetime (Figure S16). The light intensity of all sources was controlled at 1 sun, as determined by a silicon solar cell used for solar simulator intensity calibration (see Figure S17). The xenon source with a 400 nm long-pass filter (LS1) and white light-emitting diode (LED) source (LS2) had more deep blue or deep ultraviolet (UV) light, while the plasma source with emission filtered through a 450 nm long-pass filter (LS3) had a stronger infrared component. Filtering deep UV simulates final PV modules with UV absorbers (e.g., ZnO coating). We also evaluated whether the spectra of these light sources affect the stability of the solar cells. Our study demonstrated that UV in the solar spectrum does indeed accelerate the degradation of perovskite devices, but filtering UV from the solar spectrum makes the perovskite as stable as measured under white LED light (Figure S18). The degradation of the devices was similar (Figure S19), and when filtering deep UV, there was only a minor spectral difference evident in the stability of perovskite solar cells among the three light sources. The stability testing of small-area devices was performed by immersing the devices in LS2 light under open-circuit conditions. During stability testing, the devices were illuminated and heated to temperatures between 55°C and 61°C, without applying additional temperature control.
Figure S20 shows typical J-V curves of devices after different durations of light soaking, and Figure 4D displays the average PCE of control and target devices as a function of time. For the control devices, the PCE decreased to 90% of its initial efficiency (T90 lifetime) after 1,890 hours of light soaking. In comparison, the T90 lifetime of the target devices under light aging increased to 3,010 hours. Previous studies concluded that stability testing under VOC conditions is a more stringent stability test than testing under maximum power point (MPP) conditions because accumulated photogenerated excess charges accelerate perovskite degradation through enhanced ion migration. To verify this point for the perovskite composition used, we tested the same batch of control devices under both MPP and VOC conditions and found a significantly longer T90 lifetime during MPP testing (Figure S21). The thermal stability of the devices was also evaluated at elevated temperatures according to the International Electrotechnical Commission (IEC) 61215 standard. After testing in the dark at 85°C for approximately 1,000 hours, the PCE losses for both control and target devices were less than 1% of their initial efficiency (T99) (Figure S22).
Figure 4: Performance of small-area perovskite devices
Key Point 5: Efficiency and Stability of Perovskite Micro Modules
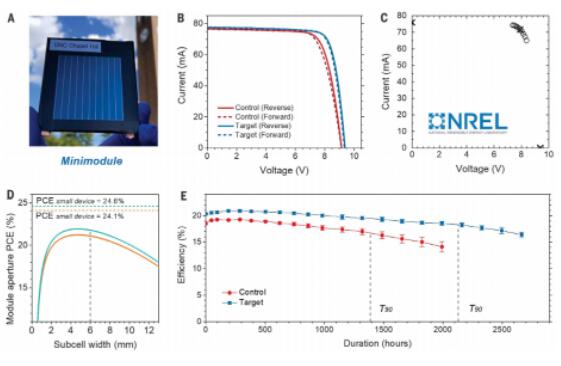
Our previous studies have shown that the non-uniform photocurrent from different sub-cells significantly reduces the fill factor (FF) of the resulting modules. The addition of BCP:PTAA improves the uniformity of the perovskite film, as evidenced by the minimal variation in device performance between batches, which should also enhance module performance. Therefore, we upgraded the small-area devices of 0.08 cm2 to micro modules with aperture areas of 20 to 30 cm2 and a geometric fill factor of 94.7% (Figure 5A). As shown in the photocurrent curves in Figure 5B, the addition of BCP enhanced the FF and VOC of the micro modules. Several micro modules were sent to the National Renewable Energy Laboratory (NREL) for PCE certification. The champion micro module with a certified aperture PCE reached 21.8%, with a J-V scan aperture area of 26.9 cm2 (Figure S25). Stable power output measurement (measuring stable photocurrent near the MPP) yielded a stable PCE of 21.1% (Figure 5C and Figure S26).
Using the recently established model, we calculated the minimum small-cell PCE required to achieve a module PCE of 21.8% (or a stable PCE of 21.1%) by assuming a fully uniform perovskite device throughout the micro module. The results are shown in Figure 5D, with further details in Figure S27. The minimum small-cell PCE required is 24.6% (stable PCE of 24.1%). This efficiency is close to the measured average PCE of the small-area devices, indicating a highly uniform perovskite film. The micro modules with BCP:PTAA HTL exhibit a VOC of 1.17 V, which is the same as the small-area devices. The photocurrent density of each sub-cell in the micro module is 23.6 mA cm-2, close to the calculated value (Figure S27A). It is noteworthy that the micro module with such a large area has a high FF of 0.803, which is also the highest reported FF for perovskite micro modules. The calculated results are shown in Figure S27B. It also demonstrates that an FF of 0.803 has reached the theoretical limit based on the achievable performance of small-area devices, indicating that the PCE loss caused by device performance non-uniformity can be neglected.
The stability of the micro modules was evaluated by subjecting five or six micro modules to light soaking under VOC conditions at an intensity of 1 sun using light source LS2. The temperature of the micro modules was measured to be between 56°C and 62°C and kept constant throughout the testing process. No higher testing temperature was used to maintain consistency with the underlying photochemistry under actual working conditions. Typical J-V curves of control and target micro modules after different durations of light soaking are shown in Figure S28. The average T90 lifetime of the micro modules with BCP:PTAA HTL reached ~2130 hours, while the T90 lifetime of the control micro modules was only ~1380 hours (Figure 5E).
Figure 5: Performance of perovskite micro modules
四、 Conclusion
The introduction of LCMs into PTAA-based perovskite solar cells has improved the efficiency, stability, and repeatability of p-i-n structured perovskite solar cells. Due to the strong interaction between the pyridine moieties in BCP and Pb2+ ions, as well as the low solubility of BCP·PbI2 in 2-ME, BCP remains at the bottom of the perovskite layer during the solution coating process. With the reduction of trap density at the bottom of the perovskite layer and the recombination of charges at the PTAA-perovskite interface, the average phase transition efficiency of BCP devices increased to 24.1%. BCP in the PTAA layer reduces the formation of amorphous layer and residual DMSO in the perovskite layer, thereby reducing the formation of voids near the grain boundaries during light soaking. BCP in PTAA also increased the T90 lifetime from ~1890 hours to ~3010 hours. The champion micro module with an aperture area of 26.9 cm2 achieved a PCE of 21.8% (stable at 21.1%).
五、Reference
Lead-chelating hole-transport layers for efficient and stable perovskite minimodules
https://www.science.org/doi/10.1126/science.ade9463
 E-mail: info@chemborun.com
E-mail: info@chemborun.com Tel: +86-574-87178138
Tel: +86-574-87178138  No. 1558, Jiangnan Road,, Ningbo, Zhejiang, China (Mainland)/31
No. 1558, Jiangnan Road,, Ningbo, Zhejiang, China (Mainland)/31